Autor

Prof. Departamento de Bioingeniería e Ingeniería Química
¿Cuál es la importancia del plegamiento de las proteínas? La estructura de una proteína está ligada a su función y permite determinar su rol y por lo tanto, los patrones que rigen a la vida. El resolver la estructura de una proteína puede tomar varios años y ser altamente costoso. Siendo esta información crucial para el desarrollo de fármacos terapéuticos y la investigación biológica, el plegamiento de proteínas es uno de los cuello de botella en la investigación científica. Recientemente, un hallazgo ha demostrado que el emplear la inteligencia artificial puede acelerar el desarrollo de nuevos fármacos y entender algunas enfermedades mediante el descifrar la estructura espacial de las proteínas, tan solo con la secuencia lineal de sus aminoácidos. El descifrar la estructura de las proteínas nos permite comprender como funcionan los procesos en la célula. Enfermedades como el Alzheimer, y el Parkinson o los priones están relacionadas al mal plegamiento de las proteínas. Si se conoce la estructura de una proteína se puede aplicar este conocimiento a la implementación de terapias o desarrollo de enzimas para funciones de interés. Sin embargo, se necesita resolver el enigma del plegamiento, para ello actualmente existen técnicas costosas como la resonancia magnética nuclear, la cristalografía de rayos X, y la Crio-microscopia electrónica.
La competencia internacional CASP “critical assesment of protein structure prediction”, se realiza desde 1994 con la finalidad de dilucidar el plegamiento de las proteínas empleando métodos computacionales. En sus primeros años, solo se han planteado métodos de interacción empleando moléculas naturales similares mediante algoritmos como los de Rossetta o aprovechar las capacidades del “docking” molecular. Sin embargo, el producto final es solo una molécula similar a una estructura natural que tiene “altas probabilidades” de ser biológicamente activa. En muchos casos luego de numerosos y costosos ensayos in vitro se obtienen un grupo de candidatos para ser analizados en modelos animales. Esfuerzos similares involucraron crear una red social para “gamificar” los algoritmos mediante aplicativos portátiles como “Folding at Home” y Foldit. Estas son maneras de emplear las redes sociales y “gamificar” los algoritmos para acelerar la producción de nuevos modelos estructurales de proteínas empleando el poder computacional de una comunidad.
En 2018, la empresa londinense DeepMind al participar en la competencia CASP13 empleo el sistema ALPHA FOLD 2 para dilucidar la estructura de una proteína. Esta empresa inglesa no pertenece al rubro de la investigación científica biológica, pero utiliza modelos de “deep learning” (DL) para resolver problemas prácticos. En esta competición DeepMind demostró que aplicando los algoritmos DL se puede obtener una dinámica molecular automatizada para inferir los plegamientos de proteínas con un mediano costo computacional. En 2018 por primera vez DeepMind obtuvo el primer lugar en la competencia de plegamiento computacional de proteínas CASP13. En 2020 la nueva versión de DeepMind conocida como AlphaFold 2 por segunda ocasión, supero a todos los equipos en la competencia CASP14.
Predecir la estructura 3D de una proteína a partir de su secuencia lineal de aminoácidos es un problema biológico complejo que debe involucrar entender las dinámicas de interacción espacial de los aminoácidos. Si se decidiera evaluar todas las configuraciones posibles se llega a la Paradoja de Levinthal con un numero de 10300 posibles conformaciones. Lo cual sería imposible de procesar dado que se necesitaría un tiempo mayor a la edad del universo para descifrar la conformación correcta.
La propuesta de DeepMind es resolver el problema del plegamiento basado en conversión de patrones de imágenes. Se propone encontrar las distancias entre todos los aminoácidos para formar una matriz de correlación. Se obtiene una primera imagen que es un mapa de calor con todas las distancias de los aminoácidos llamada PDM (“protein distance matrix”). Un algoritmo de inteligencia artificial puede procesar los parámetros de la dinámica molecular empleando PDM y un mapa de calor (Figura 1A). Adicionalmente, se encuentran los aminoácidos cruciales en la estructura espacial empleando MSA (“multiple sequence alignment”). MSA es un alineamiento masivo en donde se comparan proteínas con una relación evolutiva. En el alineamiento, se comprueba si un aminoácido cambia (“evoluciona”) debido a una mutación y que otro aminoácido de la misma proteína suele cambiar paralelamente. Estos cambios paralelos se dan debido a que estos interaccionan físicamente en la proteína y deben cambiar juntos para mantener la estructura de la proteína (coevolución estructural). Por lo tanto, DeepMind propuso combinar las distancias globales de todos los aminoácidos mediante PDM con distogramas (Figura 1B), y al mismo tiempo identificar los aminoácidos que interaccionan físicamente identificados por MSA para inferir la estructura espacial de las proteínas. Todos estos datos se analizan empleando algoritmos de redes neuronales convolucionales, usadas para reconocimiento de imágenes. El modelo “aprende” a generar una imagen como la Figura 1B; que representa la estructura tridimensional de la proteína (distograma). El distograma es una imagen que es una simple representación de la distancia tridimensional de todos los aminoácidos. Empleando un segundo algoritmo de inteligencia artificial llamado “descenso del gradiente” (gradient descent); se ajusta la estructura para plegarla optimizando los ángulos de torsión y los que interaccionan físicamente de los aminoácidos relacionados en coevolución.
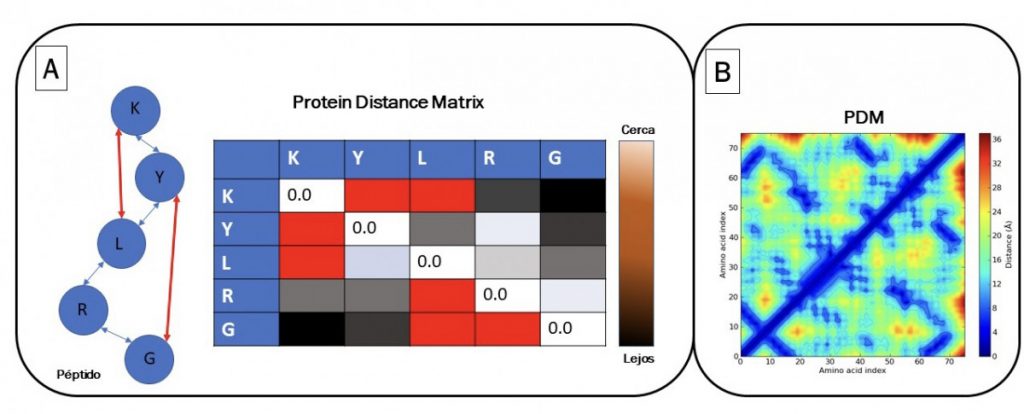
Fuente. Elaboración propia.
Para entrenar al modelo se emplean las proteínas depositadas en Protein Data Bank (PDB) que fueron generadas por técnicas más costosas como cristalografía, RMN o Electro-Crio-Microscopia (ECM). AlphaFold reconoce los patrones de plegamiento comparando datos de PDM, MSA y las estructuras depositadas de PDB obteniendo resultados altamente precisos. Para medir esta precisión se emplea el índice “global distance test” (GDT); que compara las distancias naturales con las predichas en una escala de 0 a 100, e indica el porcentaje de residuos de aminoácidos en la posición correcta [8]. Sabiendo que la cristalografía de rayos X y la RMN obtienen valores GDT de 90 o mayores, en el caso de AlphaFold 2 se obtuvo un valor de 92.4 GDT utilizando la proteína ORF8 de SARS-CoV2. Este resultado hizo que DeepMind ganara la competencia de CASP14 en 2020 (Figura 2). Si bien AlphaFold 2 aun requiere un costo computacional moderado, los valores de GDT obtenidos ya son un avance tecnológico importante y un ejemplo de como la inteligencia artificial puede resolver problemas biológicos actuales. Adicionalmente, también se plantea desarrollar “inverse protein folding”; que es una estrategia que permite deducir la “optima” secuencia lineal de aminoácidos si se conoce una estructura proteica teórica que cumple una función relevante.
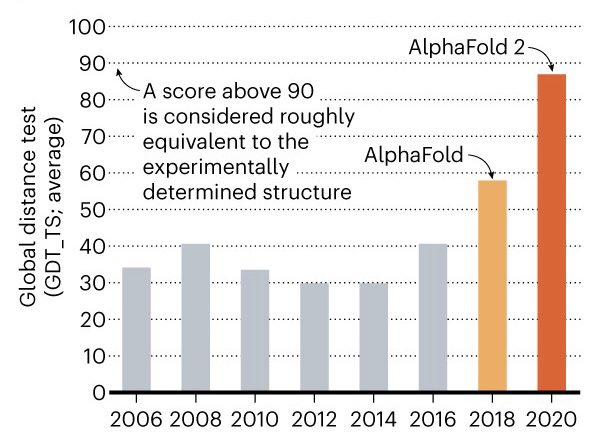
Fuente. Nature (2020).
A la fecha, la limitante al proceso de producción de proteínas de novo es la disponibilidad de estructuras cristalizadas y descritas en las bases de datos como Protein Data Bank (PDB). Únicamente con esta colección los biólogos estructurales infieren moléculas 3D similares apoyados en algoritmos de comparación espacial y así predecir una función biológica. El juego ha cambiado y AlphaFold ofrece una alternativa poderosa para predecir la estructura 3D de las proteínas y estudiar su función; por ello, se le ha catalogado como el avance tecnológico más relevante del año 2020 (Figura 3).
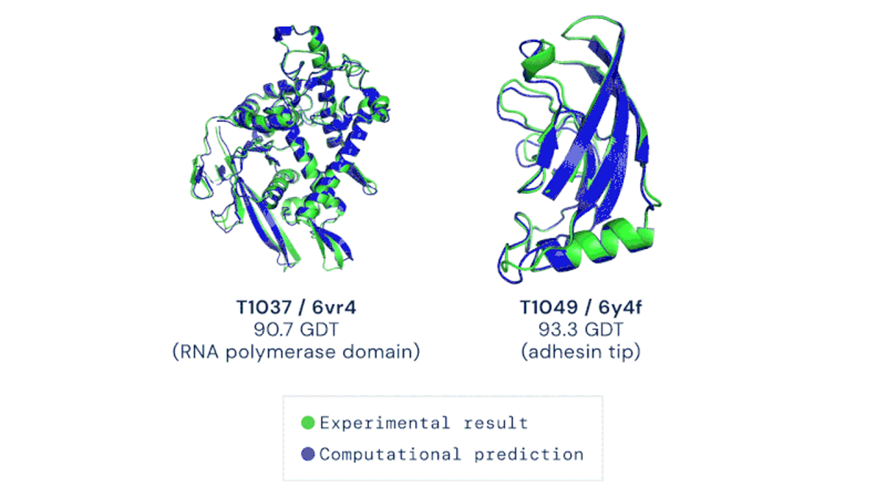
Fuente. Nature (2020).
Referencias Bibliográficas:
1.- Das Gupta D, Kaushik R, Jayaram B. Protein folding is a convergent problem! Biochem Biophys Res Commun. 2016 Nov 25;480(4):741-744. doi: 10.1016/j.bbrc.2016.10.119. Epub 2016 Oct 28. PMID: 27983988.
2.- Ewen Callaway. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 588, 203-204 (2020); doi: https://doi.org/10.1038/d41586-020-03348-4.
3.- Sikder AR, Zomaya AY. An overview of protein-folding techniques: issues and perspectives. Int J Bioinform Res Appl. 2005;1(1):121-43. doi: 10.1504/IJBRA.2005.006911. PMID: 18048125.
4.- Carol A. Rohl, Charlie E.M. Strauss, Kira M.S. Misura, David Baker. Protein Structure Prediction Using Rosetta. Methods in Enzymology. Academic Press, Volume 383, 2004, Pages 66-93, ISSN 0076-6879. https://doi.org/10.1016/S0076-6879(04)83004-0.
5.- Pande lab (August 2, 2012). «Folding@home Open Source FAQ». Folding@home. foldingathome.org. Archived from the original (FAQ) on March 3, 2020. Retrieved July 8, 2013.
6.- Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, et al. (August 2010). «Predicting protein structures with a multiplayer online game». Nature. 466 (7307): 756–60.
7.- Markoff J (10 August 2010). «In a Video Game, Tackling the Complexities of Protein Folding». The New York Times. Retrieved 12 February 2013.
8.- Senior, A.W., Evans, R., Jumper, J. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7.
9.- Levinthal, Cyrus (1968). «Are there pathways for protein folding?». Journal de Chimie Physique et de Physico-Chimie Biologique 65: 44-45.